Lightning-fast Genome Variant Detection
Web Published:
10/11/2021

Invention Summary:
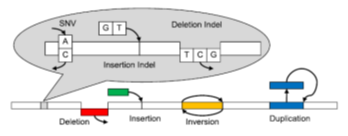
Current human whole genome sequencing projects produce massive amounts of data, often creating significant computational challenges. Different approaches have been developed for each type of genome variant and method of detection, necessitating users to run multiple algorithms to find variants.
Researchers at Rutgers University have developed the Genome Rearrangement Omni-Mapper (GROM), a novel comprehensive method of variant detection, combining mismatch, split-read, read pair, and read depth WGS evidence. GROM boasts lightning-speed runtimes an order of magnitude faster than state-of-the-art variant detection pipelines. While drastically reducing computational time, GROM detects SNVs, indels, SVs, and CNVs in a single algorithm and provides superior overall variant detection compared with commonly employed algorithms.
Advantages:
- Outperforms state-of-the-art methods (Manta, Lumpy, GATK) on validated benchmarks using annotated whole genome sequencing (WGS) data sets with Illumina short paired reads.
- Lightning-fast run times on commonly available computer hardware; more than an order of magnitude faster than other tools.
- Addresses the needs of big data analysis with superior speed, sensitivity, and precision.
- Also detects CNVs, SNVs, and indels in non-paired-read WGS libraries, as well as SNVs and indels in whole exome or RNA sequencing data sets.
Market Applications:
- Healthcare (diagnostics, stratified drug trials, personalized medicine)
- Agriculture (marker- or variant-assisted breeding)
- Research Labs and Institutions – multiple clinical applications and markets (Cancer, cardiovascular, infectious diseases, HLA typing, mendelian disorders, metabolic and immune disorders, neurological, newborn screening, prenatal screening, and preimplantation diagnostics)
Intellectual Property & Development Status:
The technology is patent pending and is currently available for research collaboration and/or licensing.
Patent Information:
| Title |
App Type |
Country |
Serial No. |
Patent No. |
File Date |
Issued Date |
Expire Date |
Patent Status |
|
|
|
ID: 2015-081
Category:
Inventors:
Keywords:
|
